

Remediating Environmental Monitoring Excursions
The release of the proposed revisions to USP General Chapter <797> in September 2021 was long awaited and came with unexpected, significant changes to Section 6, “Microbiological Air and Surface Monitoring” (see TABLE 1). Under these revisions, those who plan to perform Category 1 and 2 compounding are required to collect viable air samples at least every 6 months and surface samples at least monthly.1 Additionally, there are new requirements for Category 3 compounded sterile preparations (CSPs). Organizations that compound Category 3 CSPs are required to sample more frequently than those that compound Category 1 and 2 CSPs—viable air sampling is required monthly, and surface sampling must be performed weekly and at the completion of each batch.1
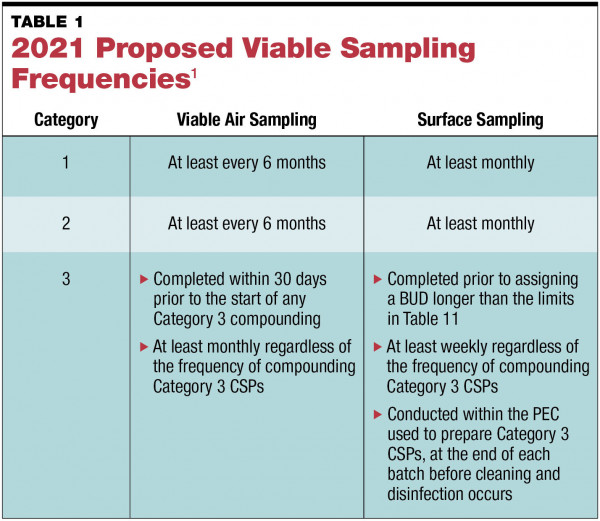
No matter the CSP category, sampling required by the proposed changes is more frequent than the current requirements of viable air sampling at least every 6 months and surface sampling periodically.2 While this increased frequency may not seem too significant, it is important to note that more frequent sampling does increase the likelihood of needing to investigate and remediate excursions. Because of this, implementation of a robust sampling program and a detailed, yet flexible, excursion investigation protocol is vital.
Excursion Guidance
The proposed version of USP General Chapter <797> does not provide specific guidance on handling microbial excursions. Rather, it provides a general foundation of the policies that must be included in standard operating procedures (SOPs). The chapter specifies the following1:
- The cause of the excursion must be investigated, and corrective action must be taken.
- Data collected in response to corrective actions must be reviewed to confirm that the actions taken have been effective.
- The corrective action plan must be dependent on the colony forming unit (CFU) count and the microorganism recovered.
- The extent of the investigation should be consistent with the deviation and should include an evaluation of trends.
- The corrective action plan must be documented.
- If levels measured during viable air or surface sampling exceed the proscribed levels, an attempt must be made to identify any microorganism recovered to the genus level with the assistance of a microbiologist.
While this guidance is helpful as a baseline, it is necessary to create an SOP that delves more deeply into next steps once an excursion is identified.
Action and Alert Levels
According to USP General Chapter <797>, the viable sampling program must include a designation of action levels to identify if an excursion has taken place. In Technical Report No. 13 (2001), the Parenteral Drug Association (PDA) defines action levels as “a level that, when exceeded, indicates a process has drifted from its normal operating range. A response to such an excursion should involve a documented investigation and corrective action.”3 USP General Chapter <797> is similar, with the additional requirement that the response must include the documented investigation.
Sterile compounding organizations typically use the action levels provided in USP <797>. In some cases, an organization may implement more stringent action levels, if the stricter levels better represent the facility’s typical state of microbial control. However, action levels cannot be less stringent than those defined in the chapter.
An organization may also choose to implement alert levels, which are defined by the PDA as “a level that, when exceeded, indicates a process may have drifted from its normal operating condition. Alert levels constitute a warning, but do not necessarily warrant corrective action.”3 It is important to note that the 2014 revision of Technical Report No. 13 does not provide action and alert level definitions, as there has been a paradigm shift to tracking incident rate, in addition to action and alert levels, in aseptic manufacturing organizations.4
Alert levels are more complicated than action levels. USP General Chapter <797> does not mention them, leaving it to the sterile compounding organization to determine if the use of alert levels is appropriate for their organization. Because the allowable action levels in the chapter are very low for both ISO Class 5 and 7 environments, alert levels for these areas are not significantly different from the action levels and do not provide a valuable indication of control drift.
Microbial Growth Identifications
The 2021 version of USP General Chapter <797> indicates that identification to the genus level is only required on air and surface samples where the number of recovered CFUs exceeds the action level. While this may reduce costs associated with testing, it may also make some individuals apprehensive about what they might be missing by not identifying every recovered colony. A best practice recommendation is to identify all recovered growth in the ISO Class 5 primary engineering control and, if an organization sees fit, identify all recovered growth to the species level. The decision to go beyond USP requirements should be risk-based, with organizations considering the CSP categories they compound, whether they do nonsterile to sterile compounding, batch size, how much anticipatory compounding is done, garbing practices, facility design and functionality, and overall workflow.
Creating an Excursion Protocol
Once an excursion is identified, next steps depend on several factors as investigations are unique to every excursion type and every organization. Sometimes the data will indicate that immediate corrective action is necessary. In other cases, it may make sense to simply monitor the situation without taking any action until the organization has a better understanding of the cause of the excursion. For example, an organization’s actions will be very different if all air samples in the buffer and anterooms exceed the action level versus having only one exceeded action level on an air sample in the anteroom.
This variability makes it difficult to create an algorithm that can handle every possible scenario. Sterile compounding organizations can create general action plans, but there must be an avenue for the responsible individuals to make appropriate decisions based on the number and types of microorganisms recovered. This is best done with the support of a microbiologist experienced in cleanroom viable sampling. SOPs must allow for flexibility in the investigation.
Documenting the Investigation
Documenting the excursion investigation creates the necessary paper trail to prove to inspectors and surveyors that the excursion was properly addressed. Documentation can occur via a paper-based system or an electronic data-management system. In either case, there should be an established form that is part of the organization’s SOPs. Sampling data, trending reports, and identification results can be attached to the investigation form to provide the necessary data without having to rewrite everything into the investigation form. As part of a thorough investigation, the following should be documented:
- Date the investigation was opened
- Identification of individual that opened the investigation
- Exceeded level sample locations, including date sampled
- Whether a single location or multiple locations had exceeded levels
- Type of excursion (action or alert level or other result that prompts action as determined by the organization)
- Notification of appropriate parties (management, state board of pharmacy, department of health, etc)
- Review of the laboratory report or internal documentation indicating inadvertent contamination of the sample
- Analysis of results and identifications
- Immediate corrective actions
- Review of any documentation or records that could provide insight into the investigation (eg, certification reports, temperature and humidity data, pressure differentials, cleaning logs, facility video records)
- Staff interviews
- Corrective action plan, including how beyond-use dating will be managed
- Results of corrective actions
- Verification of a return to a state of microbial control
It is critical that the investigation and remediation process be a team activity. Utilize the knowledge and experience of pharmacists, technicians, and infection control staff in your organization. Also consider involving a pharmaceutical microbiologist experienced in viable air and surface sampling to assist with more difficult investigations.
Case Study
A sterile compounding pharmacy at a small hospital has a 3-room cleanroom suite in which Category 2 CSPs are prepared and a segregated compounding area for nonhazardous Category 1 preparations. They outsource their viable air and surface sample collection to a certification company, who in turn outsources incubation and analysis to a contract microbiology lab. Air and surface samples are collected monthly.
The last sample session yielded an exceeded action level in the hazardous drug (HD) buffer room. The air sample collected by the door from the HD buffer room to the anteroom yielded 12 CFU/m3. The growth was identified to the genus level: 2 Staphylococcus species, 1 Micrococcus species, and 9 Corynebacterium species. Sarah is the newly appointed designated person responsible for viable sampling and investigating excursions. Following the organization’s SOPs, she starts documenting the investigation.
Sarah notes the date she is opening the investigation and that she is the person completing the documentation. She also documents the location of the exceeded level and the date the sample was collected. She indicates that this was the only exceeded action level from the sampling session performed on this date. Sarah notifies her manager of the excursion.
Before taking any corrective actions, she reviews the lab report and the sample collection documentation from the certification company for any indication that the samples were mishandled or contaminated. Although nothing is noted, she contacts the lab to be sure there were no issues during sample receipt or incubation. She is also unsure of the source of Corynebacterium species.
The lab confirms that the samples were not compromised. Because the lab is ISO 17025 accredited, they must keep record of the condition of the samples at the time of receipt and track any issues that could have compromised the samples.5 She learns that Corynebacterium species is a gram-positive rod, which is typically sourced from human skin and the nasopharynx, but may also be sourced from cattle, sheep, horses, or goats, depending on the species.6 Sarah then contacts the certification company to ensure the samples were not compromised during the sampling session. Reassured that the result is a true excursion, Sarah documents her calls with the lab and the certification company and reexamines the results.
Since she is new to the organization, Sarah reviews the HD buffer room’s viable air sample data from the last year. There have been 5 exceeded action levels in the HD buffer room, all of which have been human skin flora, as was seen in the latest sample session. The exceeded levels were not always at the same air sample location and did not exceed the action level of >10 CFU/m3/span> by more than a few colonies each time. The samples were collected under dynamic conditions, with at least 2 people in the room (typical conditions for the HD buffer room).
However, Sarah notices that the resamples—conducted to show a return to a state of control—did not occur under the same conditions because there was only one person in the room. This finding was troubling, suggesting the possibility that the room did not return to a state of control. In addition, a thorough investigation was never conducted; most of the investigations simply indicate the facility was cleaned and resampled.
Since it has been 2 months since the last excursion and the exceeded level was only 2 CFU over the action level, Sarah decides to maintain the current beyond-use dates in the HD buffer room and documents this. However, she is going to investigate the excursion more thoroughly than has been done in the past.
Sarah reviews the last two certification reports, temperature and humidity data, pressure differentials, and cleaning logs for any clues. Everything checks out as expected. This prompts Sarah to schedule some time to observe work practices and talk with staff members. Instead of simply watching work practices through the observation window, Sarah garbs and enters the HD buffer room, wearing the HD gown over the non-HD gown. While in the HD buffer room, she finds the room to be warm and notices that the two staff members are visibly sweating. Taking a few minutes to chat with staff, she learns that the room is usually uncomfortably warm. With this new information, Sarah revisits the temperature and humidity logs for the room.
The temperature and humidity in the cleanroom suite are monitored by an automated system, managed by the facilities team. The recorded temperatures are averaging 65°F, well below their goal of 70°F. Sarah looks back at the certification report, which indicates that a temperature reading taken in the HD buffer room with a calibrated thermal anemometer was 72°F. Sarah contacts the facilities team and discovers that the system temperature probes have not been calibrated for the last 2 years. This has resulted in the room appearing to be cooler than it really is.
Sarah identifies the root cause as uncalibrated temperature monitoring probes. The warm room and the two gowns made for an uncomfortable work environment, resulting in staff sweating and the release of some deeper skin flora, including the Corynebacterium species.
With the identification of the root cause, a multi-step corrective action plan is developed:
- Sarah immediately schedules the certification company to sample all viable air locations in the HD buffer room, under both static and dynamic conditions, with two staff members present. The static sampling provides data regarding the overall functionality of the room and rules this out as a cause. The dynamic sampling serves to determine if the exceeded levels are a consistent issue. Since the organization already completed several daily cleans and a monthly clean prior to receiving the initial results, additional cleaning is not deemed necessary prior to sampling.
- Sarah will work with the facilities team to ensure the monitoring system and the associated probes are calibrated annually. She will also receive copies of the calibration records so that any out of tolerance results for the probes will prompt further investigation.
- The HVAC system will be assessed to ensure all the rooms in the cleanroom suite meet the established temperature and humidity levels.
- Training will be conducted with staff on proper garbing and behavior. Staff will be reminded to contact the designated person in the event of any unconformable working conditions.
Upon completion of the calibrations and HVAC assessments, it is determined that the facility temperatures are again appropriate. The static samples did not yield any significant growth indicating facility issues. The dynamic samples demonstrated some human flora as expected but were below the action levels. Since the receipt of these results coincided with the scheduled monthly viable sampling, all routine samples were collected. In addition, Sarah scheduled the certification company to collect viable air samples in the HD buffer room for the next two days, to have a few more data points demonstrating a return to a state of control.
All samples from the monthly session and the additional HD buffer room samples were below the action level. To close out the investigation, Sarah completed the facility form, drafted a summary of actions taken including the efficacy of those actions, and attached copies of relevant data. The investigation form was submitted to management for a final review.
Summary
As demonstrated in the case study, things may not always be as they seem; thus, a thorough investigation is always necessary. Take the time to review all relevant data, even if it has previously been reviewed. A meaningful clue may have been missed, which might be key to identifying the root cause of the excursion. And remember, no two excursions are the same: Different counts, microorganisms, and facilities will make each investigation unique. Successful investigations take time, require input from a multidisciplinary team, and will ultimately improve your sterile compounding operation.
References
- United States Pharmacopeia. Briefing: <797> Pharmaceutical Compounding—Sterile Preparations. Updated August 2021. Accessed April 18, 2022. https://go.usp.org/l/323321/2021-08-31/5kmjwt/323321/1630464575pHSCnY6w/795_PHARMACEUTICAL_COMPOUNDING_NONSTERILE_PREPARATIONS_POST_Revised.pdf
- Pharmaceutical compounding—sterile preparations (general information chapter 797). In: The United States Pharmacopeia, 43rd rev., and the National Formulary, 38 ed. Rockville, MD: The United States Pharmacopeial Convention. 2008.
- Fundamentals of an environmental monitoring program. Technical Report No. 13. Revised. Parenteral Drug Association; 2001;55(5 Suppl TR13):i-iv,1-35.
- Revised Fundamentals of An Environmental Monitoring Program. Parenteral Drug Association, Inc.; 2014.
- International Organization for Standardization and International Electrotechnical Commission. 17025:2017 General Requirements for The Competence of Testing and Calibration Laboratories. 3rd edition. American National Standards Institute; 2017.
- Tille, PM. Bailey and Scott’s Diagnostic Microbiology. 14th edition. Elsevier; 2017.
 Abby Roth, CMQ/QE, QP503A Qualified, is the owner of Pure Microbiology, a microbiology consulting company that delivers practical solutions. Prior to establishing Pure Microbiology, Abby served as the director of microbiology for Clinical IQ, LLC, and was a quality director at a contract microbiology laboratory specializing in environmental monitoring.
Abby Roth, CMQ/QE, QP503A Qualified, is the owner of Pure Microbiology, a microbiology consulting company that delivers practical solutions. Prior to establishing Pure Microbiology, Abby served as the director of microbiology for Clinical IQ, LLC, and was a quality director at a contract microbiology laboratory specializing in environmental monitoring.
Like what you've read? Please log in or create a free account to enjoy more of what www.pppmag.com has to offer.
Recent Popular Articles




Current Issue
